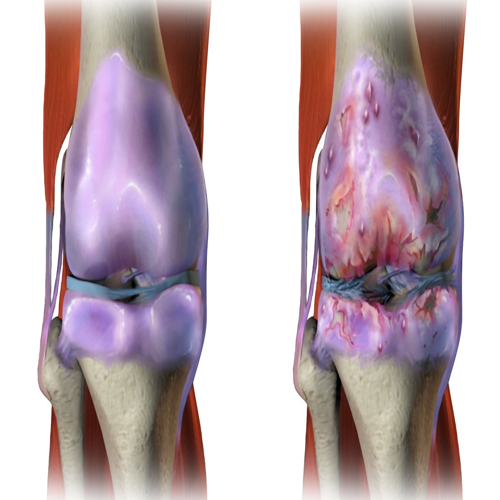
Imagine your body as a machine that repairs itself brilliantly—until one day, those same repair tools begin to harm rather than help. This is the unsettling reality behind many late-life diseases, including osteoarthritis. While osteoarthritis is often thought to arise from wear and tear, a deeper evolutionary and biological story suggests otherwise. Recent work by Dr. David Gems, a biogerontologist at University College London, challenges the traditional thinking about osteoarthritis and reframes it as a consequence of how evolution has shaped the ageing process.
By exploring this fresh perspective, we gain a new lens through which to understand why this condition emerges in older adults and how our own biology sets the stage for its development.
Reimagining Ageing: From Passive Breakdown to Programmed Overdrive
For decades, scientists assumed ageing was simply a slow collapse—a passive wearing down of our cellular machinery. That view emphasized damage, like the effects of free radicals or mitochondrial decay. However, Gems and others propose a more active culprit: programmed processes that continue running after they should stop. These processes once helped us grow, repair, and reproduce, but in later life, they overstay their welcome.
This idea falls under what’s called "programmatic theory"—the view that ageing is driven by quasi-programs. These are not adaptive features like puberty or healing, but rather developmental or regulatory processes that persist without purpose, eventually causing harm. As Gems puts it, “Senescent changes are specified by the genome, as earlier life traits are, but differ from them in being non-adaptive.”
He suggests that such programmed changes can prime tissues like cartilage for destruction, particularly in joints, where chondrocytes—the cells responsible for cartilage maintenance—start behaving abnormally late in life.
The Multifactorial Model: Ageing as a Team Effort
Osteoarthritis doesn’t emerge from one single cause. Instead, it arises from a collaboration of forces—both internal and external. Gems outlines a "multifactorial model" that splits these into two categories: disruptions and programmatic changes.
Disruptions include injuries, infections, and environmental insults. These affect people regardless of age. Programmatic changes, however, are embedded within our genome and tend to unfold only in the latter part of life. They create fertile ground for disruptions to take root and cause disease.
Take, for instance, an old knee injury. In youth, it heals without consequence. But decades later, as biological ageing alters the internal environment of the joint, that same injury may return as a chronic problem. The analogy Gems offers is striking: “Though pulling the trigger is the cause of the gun firing, the major cause of the gunshot is the cartridge.” Ageing loads the gun; the injury pulls the trigger.
Osteoarthritis is Tied to Biological, Not Just Chronological, Age
One of the most compelling arguments Gems makes is that osteoarthritis is not tied to time in the traditional sense. Rather than being a function of how many years have passed, it is better understood through the concept of biological age—how far along an organism is in its programmed lifecycle.
In a variety of mammals, OA emerges in proportion to lifespan. Mice, which live around 18 months, start showing signs of OA at around 9–12 months. Humans, who often live past 80, tend to develop OA in their 50s or 60s. This pattern repeats in rabbits, monkeys, even dolphins. It indicates that osteoarthritis is not just a matter of joints "wearing out" over time, but of genetic programs reaching a critical stage of expression.
Thus, OA appears to be “a disease of evolution,” as Gems puts it—one shaped more by the body's internal timing than by external factors alone.
Chondrocytes: From Builders to Unintended Destroyers
Much of osteoarthritis research focuses on chondrocytes, the specialized cells that build and maintain cartilage. As we age, these cells undergo dramatic changes. Traditionally, these changes were described as "cellular senescence"—a state where cells stop dividing and start secreting inflammatory molecules. But Gems argues this label is outdated and misleading.
He notes that chondrocytes in OA show behavior similar to those seen in development and tissue repair. They become hypertrophic, enlarge in size, and begin to remodel tissue. While this process is essential for bone growth in early life, in late life it becomes harmful when reactivated unnecessarily.
“It has been proposed that the accumulation of senescent cells causes OA,” he writes. But these so-called “senescent” cells may not be damaged or defective. Instead, they may be executing a once-useful program in the wrong context.
Misplaced Healing: Remodeling Gone Rogue
What if the changes in ageing cartilage aren’t just passive degeneration, but active remodeling—similar to what happens during bone fracture healing? Gems explores this possibility by examining how cartilage chondrocytes may switch on an old script: endochondral ossification, the process where cartilage turns into bone during development.
In OA, chondrocytes begin expressing genes and proteins normally associated with bone formation. These include collagen X, MMP-13, and alkaline phosphatase—molecules that help dismantle cartilage and lay the groundwork for bone growth. Gems describes this as a quasi-program: not an exact replay of development, but a confused attempt at it. He calls it “illegitimate hypertrophic differentiation.”
The cartilage begins to mineralize, and bone spurs (osteophytes) grow. These changes don’t occur randomly; they follow an orchestrated but inappropriate plan, one the body no longer needs—but can’t shut off.
Evolutionary Trade-offs and the TGF-β Switch
A central question remains: why would evolution allow such destructive programs to persist? The answer lies in the concept of antagonistic pleiotropy. This occurs when genes offer early-life benefits but cause late-life harm. Natural selection tends to favor survival and reproduction in youth, not health in old age.
One example is the TGF-β signaling pathway, which plays a key role in bone and cartilage regulation. In youth, it helps maintain cartilage by blocking hypertrophy. But as we age, this control mechanism weakens. Mice lacking Smad3, a downstream molecule of TGF-β, show increased cartilage destruction and osteophyte formation. This suggests that the loss of an “off switch” allows quasi-programs to activate unchecked.
Gems speculates that this switch was never maintained by evolution because it conferred no benefit past reproductive age. As he notes, “Such a switch could be absent in later life due to the selection shadow.”
Targeting mTOR: Can OA Be Prevented, Not Just Treated?
The mTOR pathway is another major player in this story. Known for promoting growth and cell activity, mTOR is increasingly recognized as a driver of ageing-related diseases. Chondrocytes in OA show elevated mTOR activity and decreased autophagy—a cleanup process essential for cell survival.
Drugs like rapamycin, which inhibit mTOR, have shown potential in slowing OA in animal models. However, Gems stresses that these treatments may only work if given early—before the damaging quasi-program gains momentum. Once the joint is already remodeled and cartilage lost, turning off the program may be too late.
That’s why timing is crucial. “Treatments that block chondrocyte hypertrophy or kill hypertrophic chondrocytes will have little effect on OA that has already developed,” he writes.
OA as a Cascade: Quasi-Programs All the Way Down
Gems builds a picture of OA not as a single program gone wrong, but a cascade of interlocking ones. Systemic inflammation, driven by ageing or the gut microbiome, can act as an upstream trigger. This inflammation primes joints for remodeling, which in turn causes mechanical changes, fueling further inflammation in a feedback loop.
Every component—obesity, diabetes, genetics, even menopause—feeds into this web of signals. Yet at the center lies a chondrocyte, running a script that once built bones and now destroys joints.
The Deep Cost of Evolution
Evolution never intended us to live this long. Most of the programs that damage us in old age evolved for survival in young age. “OA, like other afflictions of old age, is a disease of evolution, fully explicable only in terms of evolutionary medicine,” Gems explains.
And that’s the power of this model. It explains not just what goes wrong, but why the body allows it to happen. By tracing the roots of OA back to developmental biology, wound healing, and evolutionary trade-offs, Gems shows us that the seeds of joint destruction were planted at birth.
Understanding OA as a programmatic disease opens the door to new therapies—ones that don’t just fix damage, but prevent the script from being read in the first place.
The study is published in the journal Osteoarthritis and Cartilage. It was carried out by David Gems from University College London.